In the annals of history, the quest for material design was a formidable endeavor. For over a millennium, ambitious alchemists experimented with combinations of lead, mercury, and sulfur in a futile attempt to conjure gold. Renowned figures such as Tycho Brahe, Robert Boyle, and Isaac Newton were among those who ventured into the perplexing world of alchemy, striving for a breakthrough that eluded them.
Fast forward to today, and materials science has evolved significantly. For the last 150 years, researchers have leveraged the periodic table of elements, gaining insights into the distinct properties of different materials and debunking the notion of transforming one element into another. Moreover, recent advancements in machine learning have greatly enhanced our ability to analyze the structure and properties of various molecules and substances. A groundbreaking study led by Ju Li, the Tokyo Electric Power Company Professor of Nuclear Engineering at MIT and a professor of materials science and engineering, heralds a new era in materials design. The findings of this research are detailed in the December 2024 issue of Nature Computational Science.
Currently, many machine-learning models utilized for characterizing molecular systems are founded on density functional theory (DFT). This quantum mechanical approach calculates the total energy of a molecule or crystal by examining electron density distribution—the average number of electrons found in a specified volume around a point in space near the molecule. Walter Kohn, a co-inventor of this theory, was awarded the Nobel Prize in Chemistry for his contributions 60 years ago. While DFT has proven effective, it is not without limitations. According to Li, “Its accuracy can be inconsistent, and it primarily reveals only the lowest total energy of the molecular system.”
The Solution: “Couples Therapy” for Molecular Properties
Li’s team has adopted an alternative computational chemistry technique known as coupled-cluster theory, or CCSD(T), which is often regarded as the gold standard of quantum chemistry. Though the accuracy of CCSD(T) calculations significantly surpasses that of DFT, it comes with its own set of challenges. Li notes that performing these calculations is computationally intensive—doubling the number of electrons in a system can make computations 100 times more cumbersome. Consequently, CCSD(T) has typically been restricted to small molecules, comprising around ten atoms.
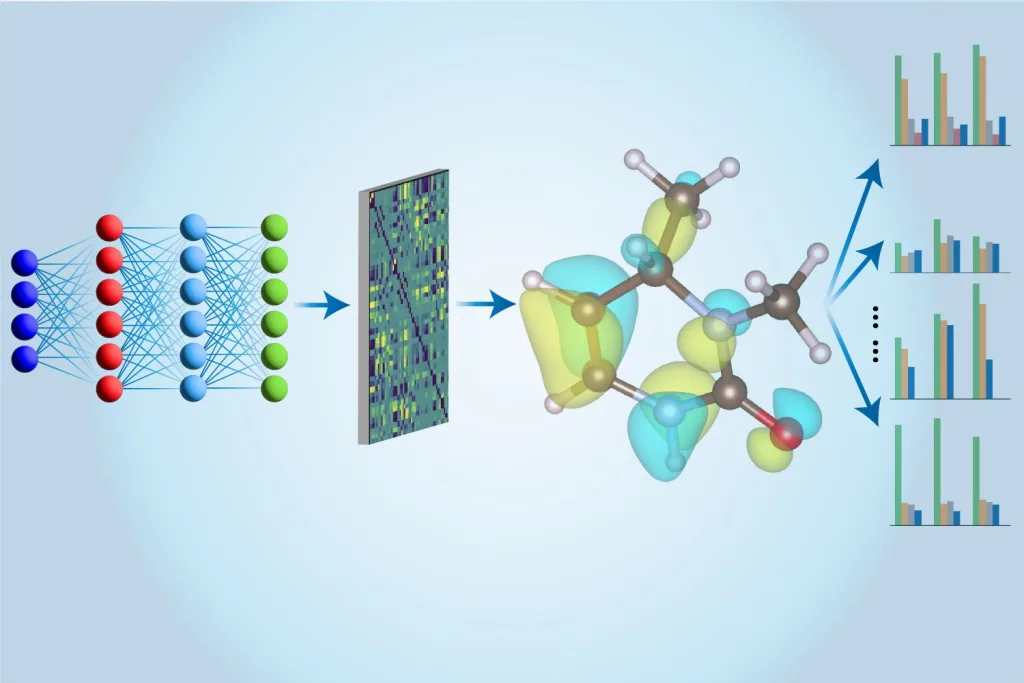
This is where machine learning makes a monumental impact. Initial CCSD(T) calculations are conducted on traditional computers, and these results are then used to train an innovative neural network developed by Li and his team. Once trained, the neural network can execute these calculations much more rapidly by employing approximation techniques. The beauty of this neural network model lies in its ability to extract a wealth of information about a molecule beyond just its energy. As MIT PhD student Hao Tang puts it, “In prior studies, multiple models were used to evaluate various properties. Our approach employs a single model to assess all these aspects, hence we refer to it as a ‘multi-task’ methodology.”
The “Multi-task Electronic Hamiltonian network,” or MEHnet, uncovers various electronic properties, including dipole and quadrupole moments, electronic polarizability, and the optical excitation gap—essentially, the energy required to elevate an electron from its ground state to its first excited state. Tang explains that, “The excitation gap plays a crucial role in a material’s optical properties, determining the frequency of light absorption by a molecule.” An added advantage of their CCSD-trained model is its capability to elucidate properties of not only ground states but also excited states, along with predicting the infrared absorption spectrum, which relates to a molecule’s vibrational characteristics.
The strength of their approach largely stems from the network architecture. By leveraging the expertise of MIT Assistant Professor Tess Smidt, the team employs an E(3)-equivariant graph neural network, whereby the nodes signify atoms, and the edges connecting them represent atomic bonds. Furthermore, customized algorithms that integrate fundamental physics principles into the model facilitate accurate assessments of molecular properties based on quantum mechanics.
Verification Process
When tested against known hydrocarbon molecules, Li’s team’s model outperformed DFT alternatives and closely aligned with experimental results documented in existing literature.
Qiang Zhu, a materials discovery expert at the University of North Carolina at Charlotte (not affiliated with this study), commended their achievements, stating, “Their method enables effective training with limited data while delivering superior accuracy and computational efficiency compared to existing models. This exciting work exemplifies the powerful synergy between computational chemistry and deep learning, paving the way for innovative electronic structure methods.”
The researchers initially applied their model to small, nonmetallic elements—such as hydrogen, carbon, nitrogen, oxygen, and fluorine—which are fundamental to organic compounds. They have since progressed to investigating heavier elements like silicon, phosphorus, sulfur, chlorine, and even platinum. After training on smaller molecules, the model is adaptable to increasingly larger molecular structures. Li notes, “Traditionally, calculations were constrained to a few hundred atoms with DFT and around ten with CCSD(T). Now, we can handle thousands of atoms and, potentially, tens of thousands in the future.”
While the team is still assessing known molecules, their model holds the potential to characterize unseen molecules and predict the properties of hypothetical materials comprising different molecular combinations. Tang adds, “Our aim is to utilize these theoretical tools to identify promising candidates that meet specific criteria before recommending them for experimental validation.”
Future Prospects
Looking ahead, Zhu is optimistic about the possibilities. “This methodology could enable high-throughput molecular screening—where precision in chemical accuracy is critical for discovering novel molecules and materials with desirable attributes,” he emphasizes. Once they demonstrate the capability to analyze large molecules—possibly comprising tens of thousands of atoms—Li envisions the potential to innovate new polymers or materials applicable in drug design or semiconductor technologies. Additionally, exploring heavier transition metal elements could yield new materials for batteries, an area currently under significant demand.
As Li sees it, the future brims with potential. “Our ambition is not limited to one area. Ultimately, we aspire to extend our coverage across the entire periodic table with CCSD(T)-level accuracy but at a lower computational cost than DFT. This advancement should enable us to tackle a broad spectrum of challenges within chemistry, biology, and materials science. The scope of these challenges remains tantalizingly vast,” he concludes.
This research received support from the Honda Research Institute. Hao Tang acknowledges funding from the Mathworks Engineering Fellowship. Partial calculations were executed on the Matlantis high-speed universal atomistic simulator, the Texas Advanced Computing Center, the MIT SuperCloud, and the National Energy Research Scientific Computing resources.
Photo credit & article inspired by: Massachusetts Institute of Technology